金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
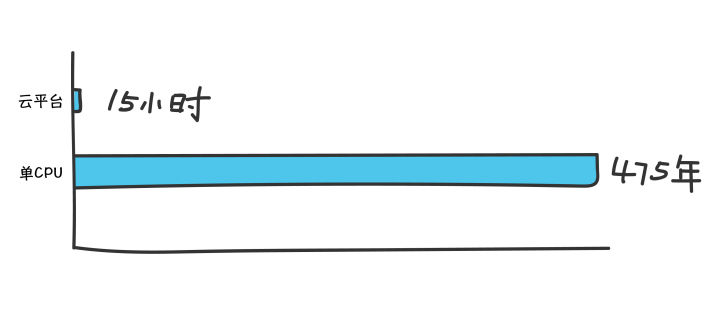
看看生信的一个方向,单细胞组学分析
这两篇文章非常相似,他们还针对大型数据集细胞注释困难的问题开发了一个工具(Snapseed),两篇文章都在用,想学习大规模单细胞数据整合的可以学习下
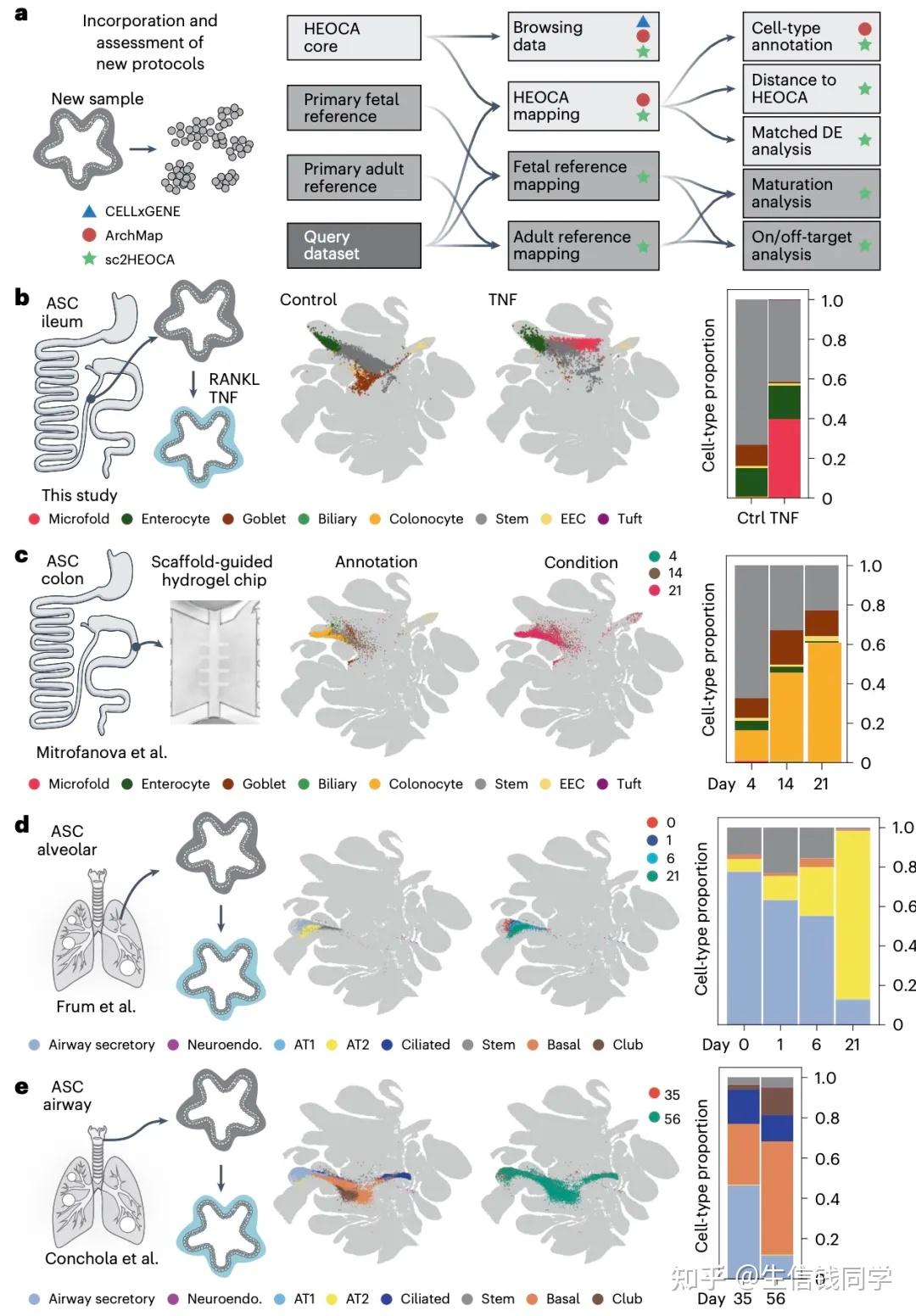
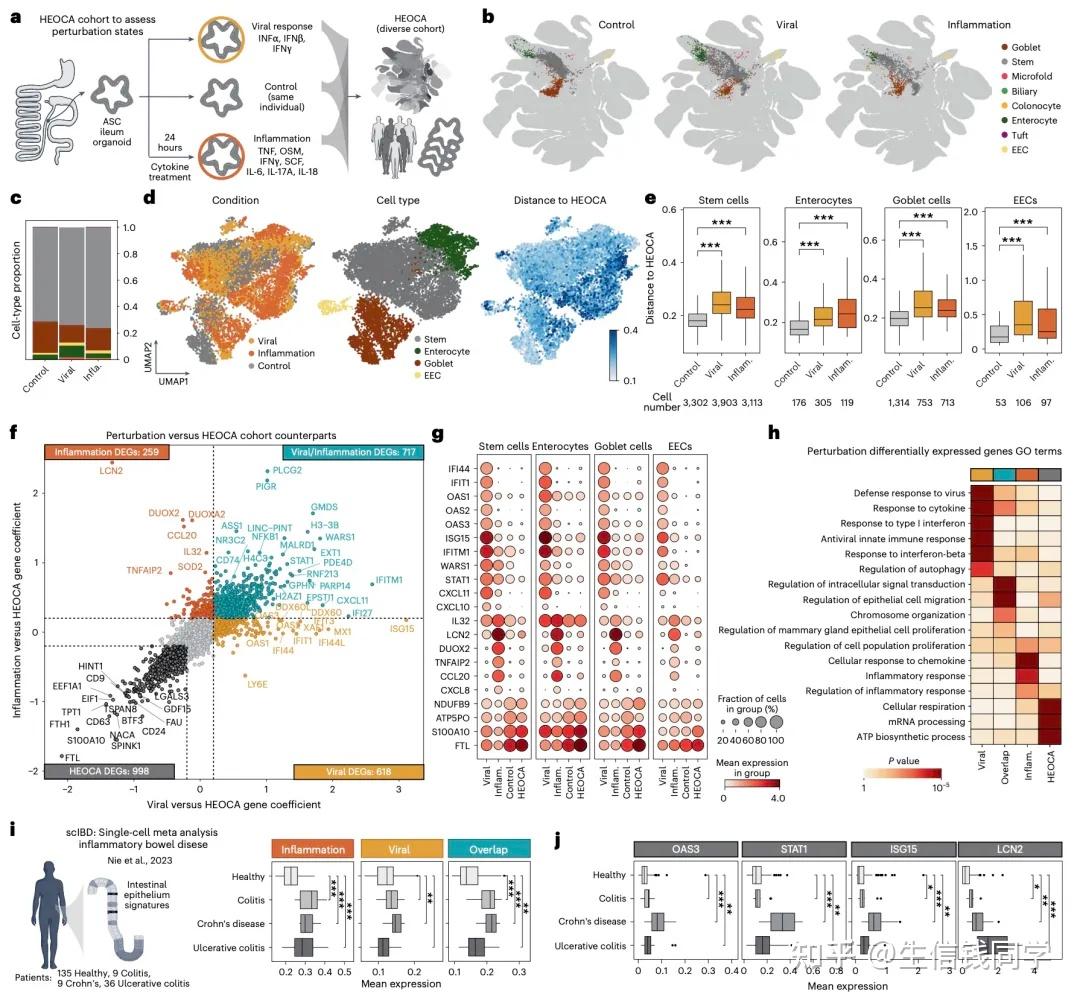
Snapseed 根据手动定义的标记基因集合对单个细胞类型或细胞类型层次进行单细胞数据集注释。加速对非常大的数据集的注释是快速而简单的。
为了注释每个细胞簇的细胞类型,研究采用了 SnapCell 方法。该方法通过计算各标记基因相对于细胞簇的受试者工作特征曲线下面积(ROC AUC)和倍数变化值(fold change)进行注释。对于特定细胞类型的多个标记基因,取其最大 AUC 值和最大倍数变化值作为代表。通过计算这些标记的平均 AUC 和平均倍数变化值来表征每个细胞类型,并基于这些标准为每个细胞簇注释最具特异性的细胞类型。


人类内胚层类器官单细胞转录组综合细胞图谱:解码器官发育与疾病的规律
跨组织类器官单细胞图谱揭示人类内胚层器官发育特征
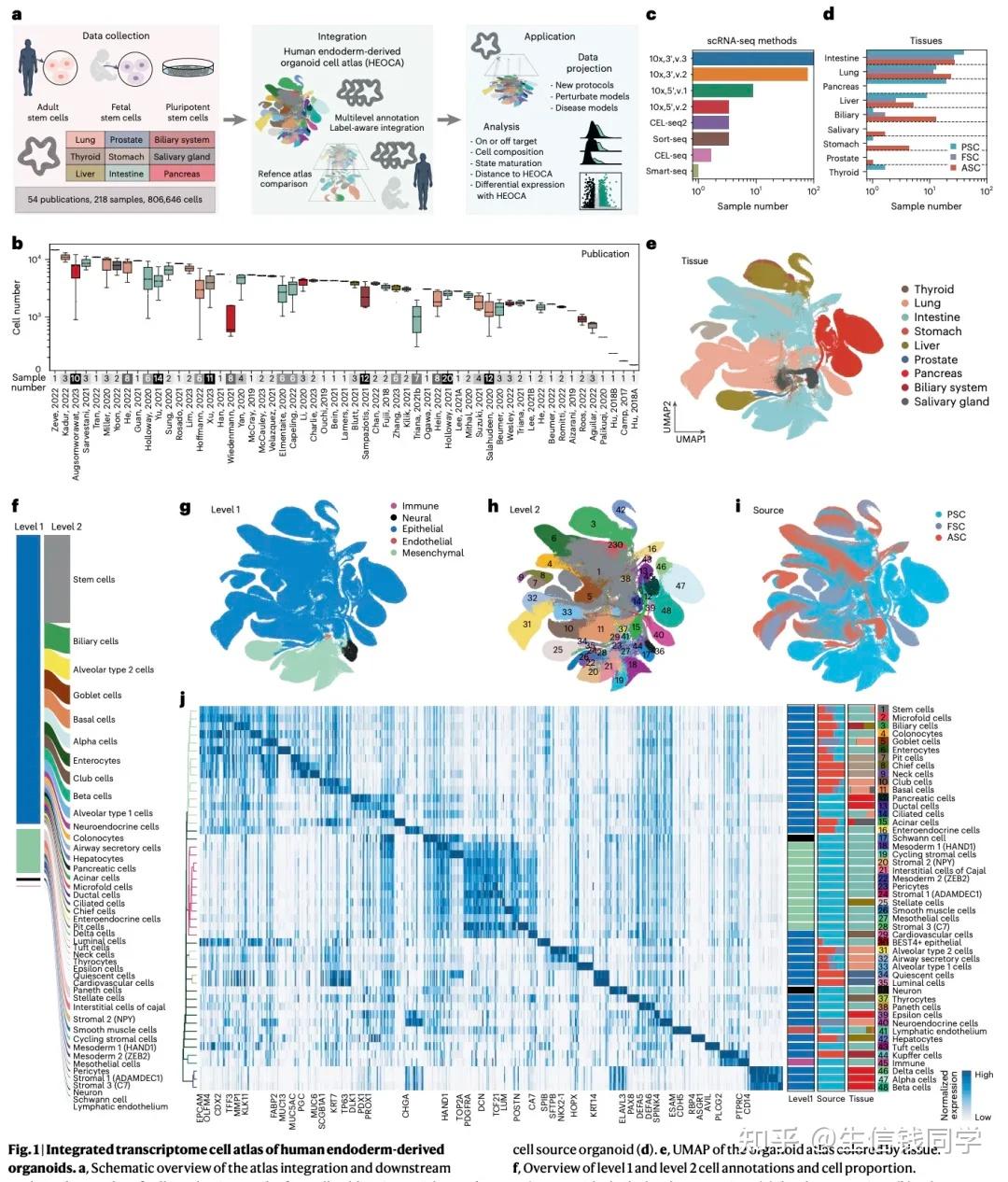
2025 年 5 月,国际顶级学术期刊《自然・遗传学》(Nature Genetics)发表了一项由瑞士罗氏制药、亥姆霍兹慕尼黑中心、巴塞尔大学等多机构合作完成的重大研究成果。研究团队通过整合全球 55 项研究的单细胞转录组数据,构建了首个人类内胚层类器官细胞图谱(HEOCA),涵盖 9 种内胚层来源器官、近百万个细胞,为理解人类器官发育、疾病建模及药物研发提供了重要的参考框架。
一、整合跨时空数据:构建类器官研究的 “基因地图”
类器官作为模拟人体器官发育的 3D 细胞培养模型,在疾病研究和药物开发中具有巨大潜力。然而,不同实验室的培养条件、干细胞来源和技术平台差异,导致类器官的细胞组成和功能难以系统比较。
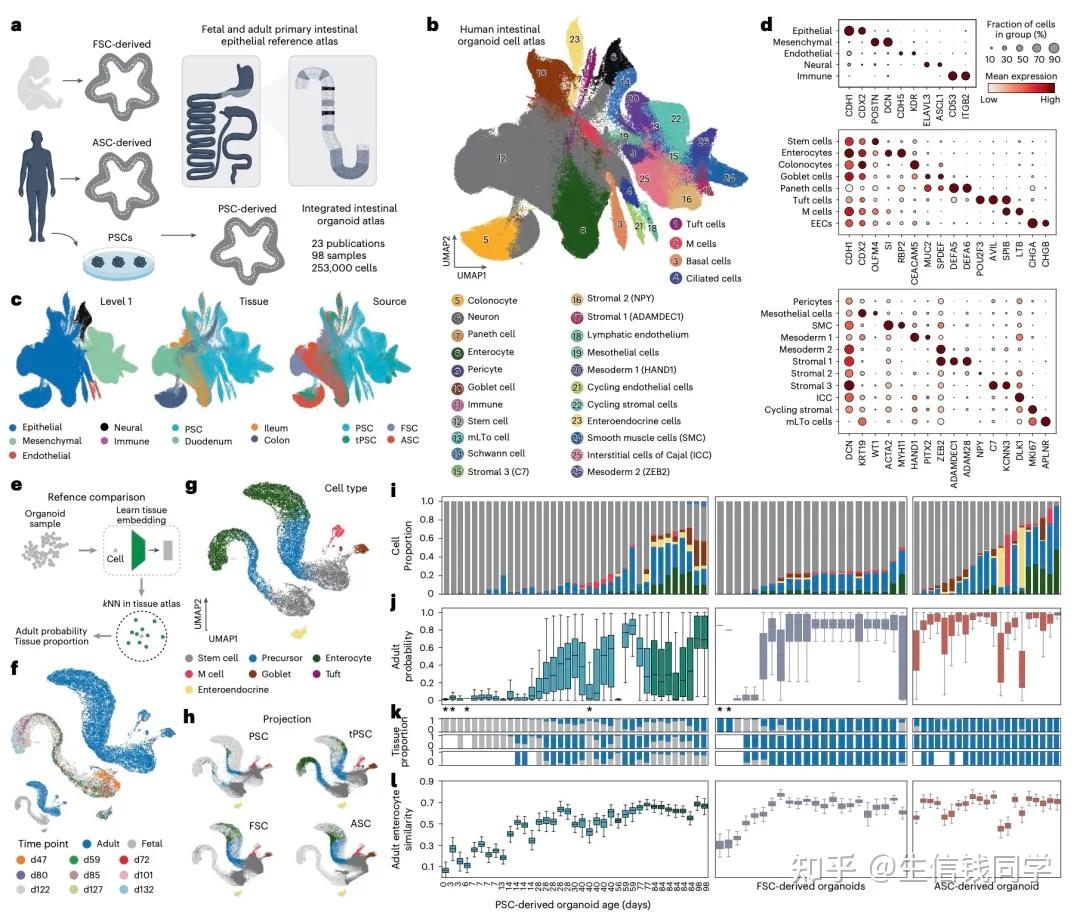
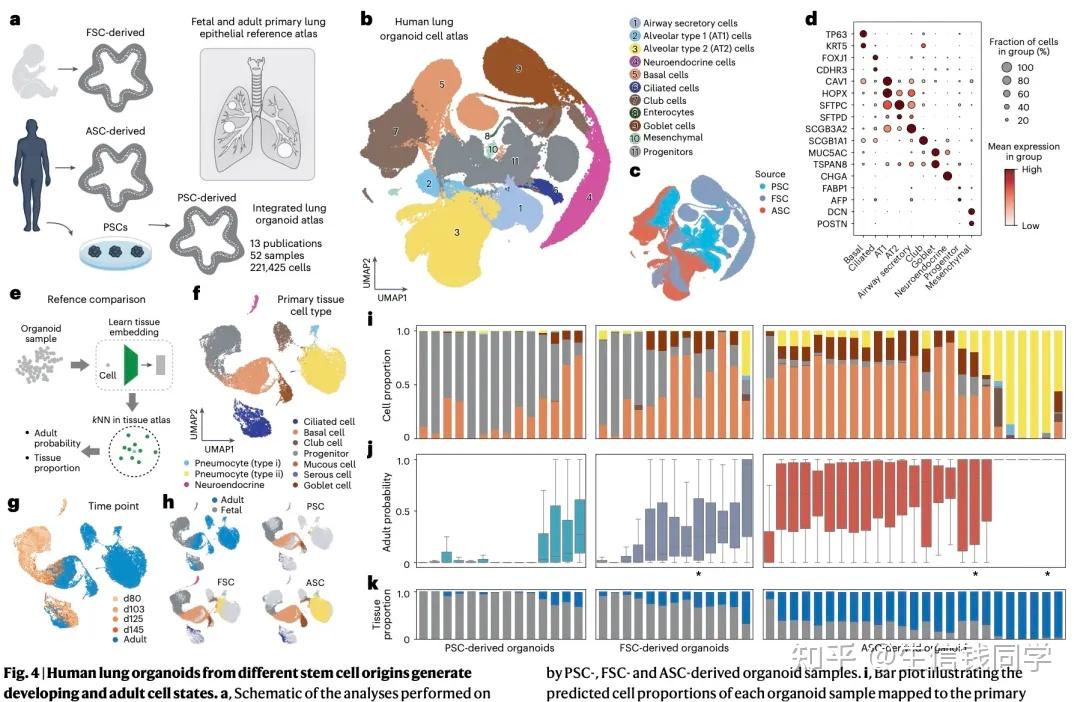
数据规模与技术创新:研究团队整合了218 个样本的单细胞 RNA 测序(scRNA-seq)和单核 RNA 测序数据,包括来自多能干细胞(PSC)、胎儿干细胞(FSC)和成体干细胞(ASC)的类器官模型,覆盖甲状腺、肺、肠道、肝脏等9 种内胚层器官。通过scPoli 算法消除批次效应,首次实现跨协议、跨组织的数据整合,构建了包含5 个细胞大类、48 个细胞类型及 51 个子类型的 hierarchical 注释体系。
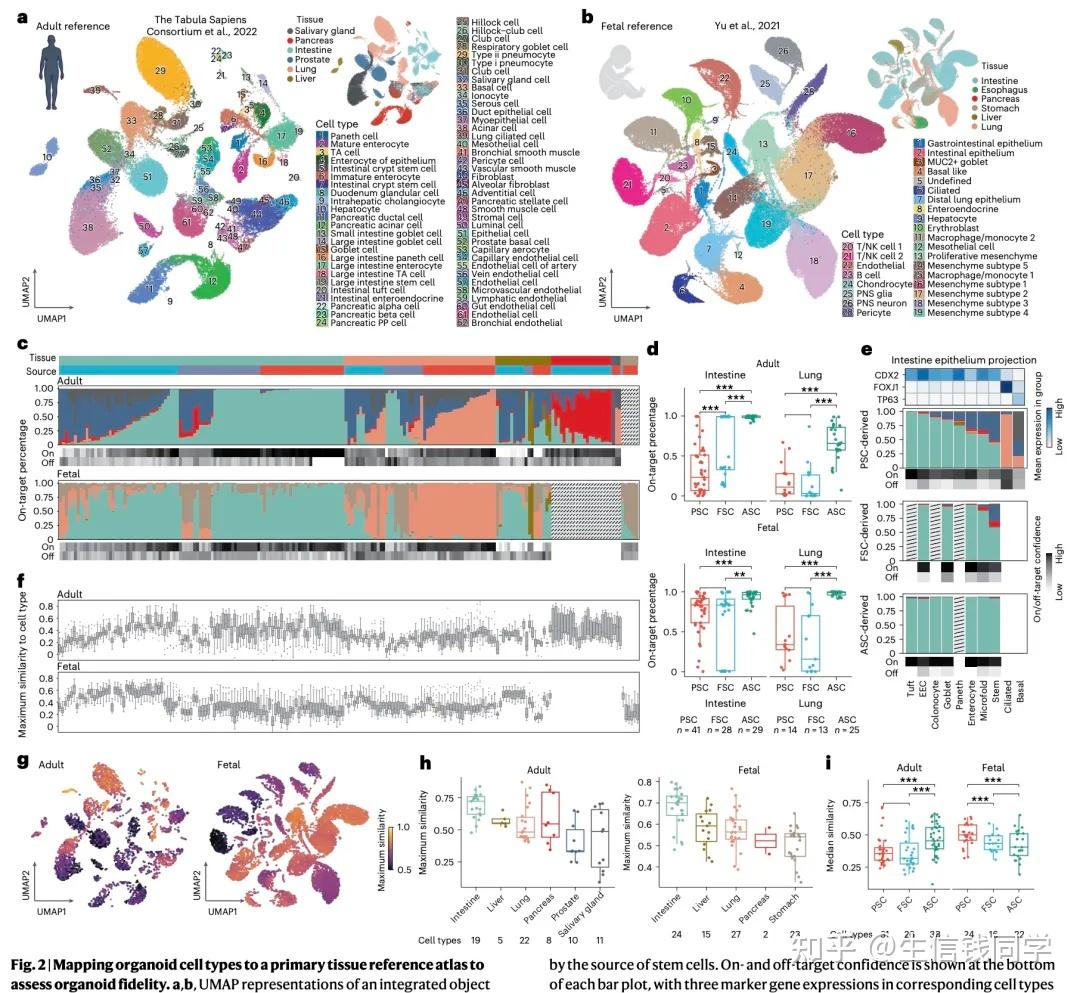
跨物种对比与 fidelity 评估:通过将类器官细胞投射到人类胎儿和成人原代组织图谱,研究发现:
ASC 来源类器官与成人组织匹配度最高(如肠道类器官达 98.14%),适合模拟成人生理状态;
PSC 来源类器官更接近胎儿细胞特征,可用于早期发育研究;
FSC 来源类器官呈现中间状态,提示其在模拟发育过渡期的潜力。

He, Z., Dony, L., Fleck, J.S. et al. An integrated transcriptomic cell atlas of human neural organoids. Nature 635, 690–698 (2024).
Xu, Q., Halle, L., Hediyeh-zadeh, S. et al. An integrated transcriptomic cell atlas of human endoderm-derived organoids. Nat Genet 57, 1201–1212 (2025).
snapseed现在已经整合到HNOCA-tools工具中了
# Human Neural Organoid Cell Atlas Toolbox 代码注释
# 功能:处理和分析人类神经类器官单细胞图谱数据
# 1. 细胞类型注释模块
import hnoca.snapseed as snap
from hnoca.snapseed.utils import read_yaml
# 读取预定义的细胞类型标记基因配置文件
# YAML格式示例: {'神经元': ['NEUROD1', 'MAP2'], '星形胶质细胞': ['GFAP', 'S100B']}
marker_genes = read_yaml("marker_genes.yaml")
# 基于标记基因对细胞簇进行快速注释
# group_name: 聚类结果字段名
# layer: 使用的表达矩阵层,通常为对数归一化后的数据
snap.annotate(
adata, # AnnData对象,存储单细胞数据
marker_genes, # 标记基因配置
group_name="clusters", # 聚类分组字段
layer="lognorm", # 使用的表达矩阵
)
# 处理多层级细胞类型注释
# 适用于复杂细胞谱系结构(如: 神经前体细胞 -> 中间神经元 -> 锥体神经元)
snap.annotate_hierarchy(
adata,
marker_genes, # 包含层级结构的标记基因
group_name="clusters",
layer="lognorm",
)
# 2. 数据映射模块 - 将新数据映射到参考图谱
import scvi
import hnoca.map as mapping
# 加载预训练的参考图谱模型
# 模型使用scANVI框架训练,支持零样本学习能力
ref_model = scvi.model.SCANVI.load(
os.path.join("model.pt"), # 模型保存路径
adata=ref_adata, # 参考数据集
)
# 创建图谱映射器实例
mapper = mapping.AtlasMapper(ref_model)
# 将查询数据集映射到参考图谱
# retrain: 部分重训练策略,平衡速度与准确性
# max_epochs: 最大训练轮次
# batch_size: 批次大小,影响GPU内存使用
mapper.map_query(
query_adata, # 待映射的查询数据集
retrain="partial", # 训练策略: full/partial/none
max_epochs=100, # 最大训练轮次
batch_size=1024, # 批次大小
)
# 基于加权KNN算法进行标签传递
# k: 近邻数,影响标签传递的平滑度
mapper.compute_wknn(k=100)
# 执行标签传递,从参考图谱到查询数据
# label_key: 参考图谱中的细胞类型字段名
celltype_transfer = mapper.transfer_labels(label_key="cell_type")
# 计算查询数据在参考图谱中的"存在分数"
# 反映查询数据与参考图谱中各类细胞的匹配程度
presence_scores = mapper.get_presence_scores(split_by="batch")
# 3. 差异表达分析模块
import hnoca.stats as stats
# 标准差异表达分析(DE)
# 比较两组细胞群体(如: 类器官 vs 体内组织)
de_df = stats.test_de(
joint_adata, # 合并后的数据集
group_key="origin", # 分组依据字段
return_coef_group="organoid", # 返回系数的组别
adjust_method="holm", # p值校正方法
)
# 基于图谱的配对差异表达分析
# 将查询数据与参考图谱中匹配的"虚拟细胞"进行比较
# 第一步: 生成匹配的表达谱
matched_adata = mapper.get_matched_expression()
# 第二步: 执行配对DE分析
de_df = stats.test_de_paired(
query_adata, # 原始查询数据
matched_adata, # 匹配的参考数据
adjust_method="holm", # p值校正方法
) |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2025-5-30 10:43
发表于 2025-5-30 10:43


 提升卡
提升卡