国内医疗器械上市前需做国内注册,医疗器械出口美国需要做FDA注册,FDA注册流程是什么样呢,今日做个简单分享。
美国医疗器械监管机构为食品药品监督管理局(Food and Drug Administration, 简称FDA)。
美国《食品、药品和化妆品法》第 201(h)节规定,医疗器械是指符合以下条件的仪器、装置、工具、机械、器具、植入物、体外试剂及其它相关物品,包括组件、零件或附件:
1.明确列在《国家处方集》(National Formulary)或《美国药典》(the Unite States Pharmacopeia)或前述两者的附录中者。
2.预期使用于动物或人类疾病,或其它身体状况的诊断,或用于疾病之治愈、减缓与治疗者。
3.预期影响动物或人体身体功能或结构,但不经由新陈代谢来达到其主要目的者。
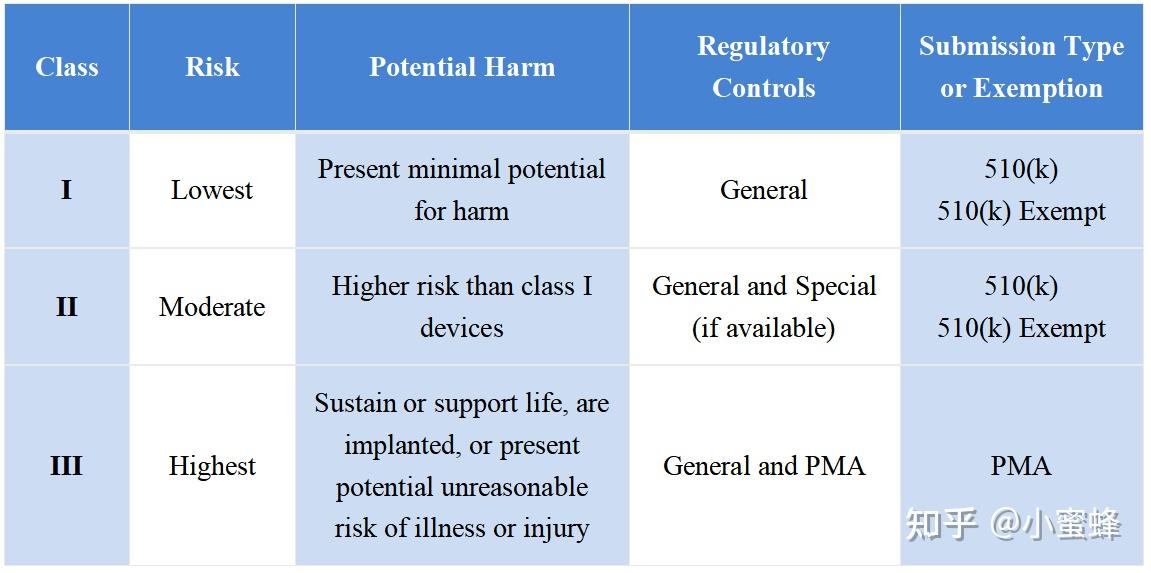
医疗器械产品分类:医疗器械根据其风险程度分为三类(I类、II类或III类)。随着器械类别从I类到II类再到III类,监管控制强度也逐步增加,I类器械受最少的监管控制,而III类器械则受到最严格的监管控制。器械类别、监管控制和提交类型总结如下表所示:

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡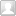 发表于 2025-7-9 18:23
发表于 2025-7-9 18:23


 提升卡
提升卡


