经常出现一种“检测悖论”:患者在检测中明确出现阳性靶点,指南也支持用靶向药,但开方或报销环节却卡在一句话上:你用的检测试剂不是这款药“绑定”的伴随诊断(CDx)。于是,患者要么自费换检,要么被迫走更慢、更贵的路径,甚至错过最佳治疗窗口。 PMDA 在 2026 年 2 月 20 日发布并上线的《非特定药物伴随诊断(Drug-agnostic CDx)适用性评估报告》,本质上就是在回应这个临床错位:当同一生物标志物的检测已经足够标准化时,监管不应继续把“药-检”用合同式的方式锁死,而应把决策锚点还给生物标志物本身。 (pmda.go.jp) PMDA 如何判断 EGFR 等成熟靶点具备 “Drug-agnostic” 的技术前提?这份评估并不是抽象宣言,而是把“可互换”拆成了可审评的条件。报告引用并沿用日本既有的 Drug-agnostic 监管框架,先给出三条“门槛型”要求:
在 EGFR(非小细胞肺癌、排除 exon20 insertion)这一案例里,报告列出了多个候选产品(PCR、NGS 面板、综合基因组分析以及软件类产品等),并将互换性讨论落到两个关键审评维度:
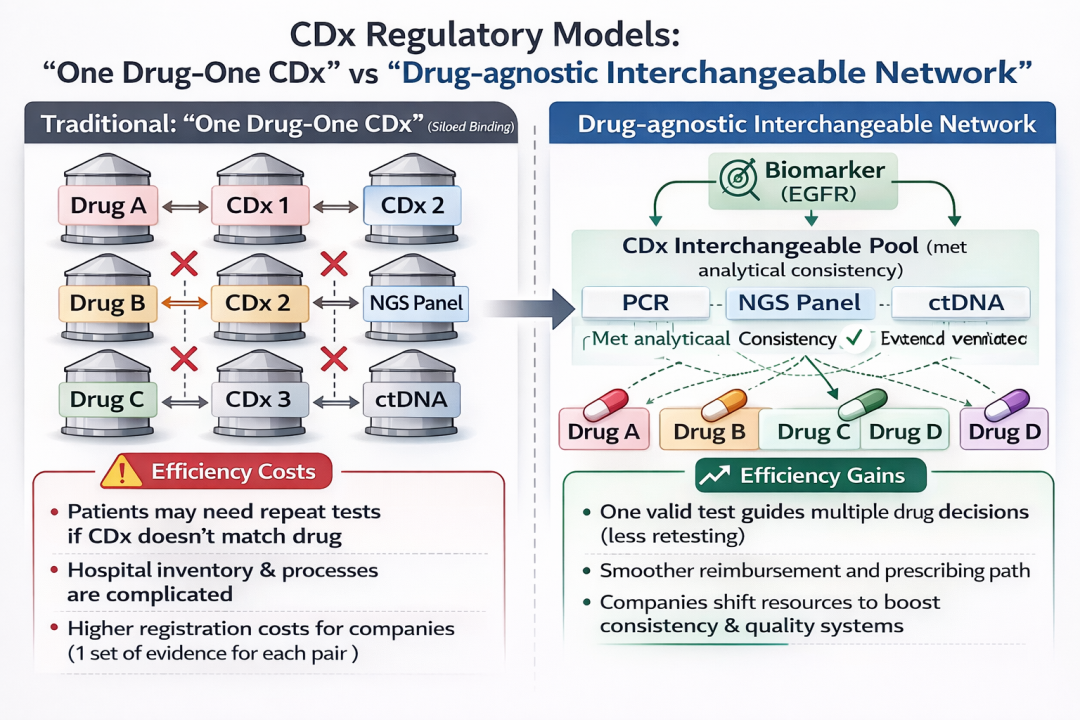
一句话总结 PMDA 的技术路线:先证明“分析一致性”足够高,再用“适用范围+标签约束”把残余不确定性管理起来。 CDx 正从药物“说明书附件”升级为精准医疗的“底层操作系统”过去的“一药一检”,监管逻辑更像是产品对产品:某药的有效性证据使用了某个检测方法,于是该检测被锁定为该药的 CDx。这个逻辑在早期靶点和检测尚不成熟时能降低不确定性,但当 EGFR 这类靶点进入“多药、多代际、多检测技术”并存阶段,强绑定会反向制造系统成本:
Drug-agnostic 的监管转向,核心不是“放松”,而是把审评锚点上移到更高维度: 监管的松绑并非门槛的降低,而是对检测一致性提出了更高维度的“标准共振”要求。 在 PMDA 的既有文件体系里,Drug-agnostic CDx 的基本思路早在 2022 年的通知与指南中就已铺垫:当可以科学论证某已获批 drug-agnostic CDx 能用于识别新治疗产品的适用人群时,部分情形下甚至不一定需要随药物申报同步去做 CDx 的“绑定式变更”。 Drug-agnostic 的本质是重还“诊断决策权”给生物标志物,而非营销契约。 对 NGS 大面板、液体活检与全球监管的外溢效应1)对 NGS 大面板:从“多药通行证”走向“可审计的一致性工程” 2)对液体活检(ctDNA):互换性不是“组织 vs 血液”的简单替代 3)对全球监管:日本可能成为“从强绑定走向可互换”的可复制样板 图名:CDx 监管模式从“烟囱式绑定”到“互换式网络” 下半场竞争拼的不是靶点数量,而是系统效率EGFR 只是一个起点。更重要的是 PMDA 用一个成熟靶点把规则讲透:当检测进入可标准化阶段,监管会从“锁定某个产品”转向“认可一套可互换的证据体系”。 伴随诊断正在从药物的“说明书附件”,升级为精准医疗的“底层操作系统”。 参考资料PMDA | Evaluation Report on Applicability to Drug-agnostic Companion Diagnostics (2026-02-20). URL: https://www.pmda.go.jp/english/rs-sb-std/rs/0027.html (pmda.go.jp) 注:本文内容仅供全球行业动态参考,不构成任何投资建议或临床医疗决策依据。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号