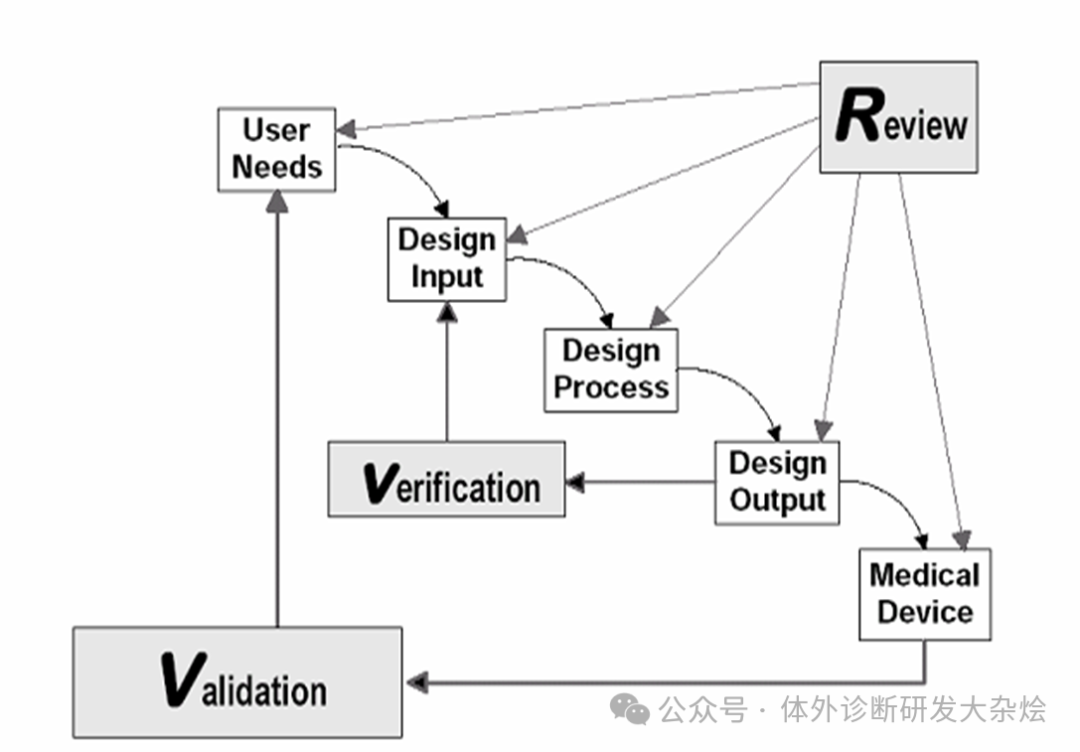
质量管理体系的基本要求:注册申请人应当按照《医疗器械生产质量管理规范》及附录的要求基于科学知识、经验以及风险管理原则,建立与产品实现过程相适应的质量管理体系,包括委托生产(如有)、临床评价(含临床试验)等环节,以确保其在医疗器械全生命周期管理过程中有效运行,保证设计开发、生产等过程数据真实、准确、完整和可追溯(反复念三遍),并与注册申报资料一致。 注册核查要求:应当结合注册申报资料组织开展注册质量管理体系核查,重点关注与产品研制、生产有关的设计开发、采购、生产管理、质量控制等内容。产品真实性核查应当全面、客观。注册核查的重中之重是真实性核查,什么叫真实性核查,通俗点讲就是是不是真的开发了,开发的产品符合要求了,能放大转产了,能通过相应的质量标准了。 接下来是本次的重点内容,假如你是一名试剂盒开发技术人员,应该怎么样留存研制记录,以应对将来的核查呢? 原始研发记录:除直接输出的主体试验数据(原始研发记录(原材料、工艺及反应体系研究数据)、分析性能数据、稳定性研究数据等)外,还应当保留设计开发过程中的辅助记录,如物料采购、领用记录及使用台账、仪器设备原始数据及相应使用记录、称量记录、配制记录、样本库信息等。开展临床试验的,应当保留临床试验过程有关的试验器械(试剂)出库记录、储运记录、回收处置记录等,让试剂开发到注册的动作皆有迹可循。 设计开发记录:此文件主要记录了整个试剂盒经历的各个重要阶段,包括设计开发策划、输入、输出、评审、验证、确认、转换、变更,以及设计开发过程中建立的记录,确保历次设计开发最终输出过程及其相关活动可追溯。
设计开发策划:应包括项目选择(市场需求等),可行性分析(法规、技术、经济等),项目立项,风险管理计划、报告等。 设计开发输入:一般包括法律法规、国家标准、行业标准、国内外指南文件、标准品或者参考物质信息、用户需求、产品适用范围、前代或者同类产品的技术指标、产品风险、标准草案、产品预研工艺、主要原料信息、设备信息、参考文献等。 设计开发输出:应当满足输入要求,以及符合用户需求和产品设计需求,应当关注产品适用范围、功能性、安全性、有效性、质量可控性。具体相关文件应包括物料清单、设备清单、SOP、工艺文件、检验规程及标准、说明书、技术要求、包装标签样稿等。 设计转换(试生产3批):应当保留产品设计转换活动的所有记录,以表明设计和开发输出成为最终产品规范前已得到充分验证且适用于常规生产,并确保生产工艺在使用确定的原材料和设备条件下,持续稳定生产出符合预期用途和产品技术要求的产品。通常设计转换会连续生产三批次产品,至少使用两批次不同原料。这三批产品后续可以用于注册检测以及临床。 设计确认(临床试验):应当按照临床试验方案及合同履行相应职责,并保存相关文件和记录(临床使用试剂盒整个周期记录,如出库记录、储运记录、回收处置记录,临床样本收集信息,检测原始记录等)。需强调的是临床试验产品为已经定型产品。 体考最终我们会得到4类结果:“通过核查”、“未通过核查”、“整改后通过核查”、“整改后未通过核查”。希望大家都能“通过核查”。 |


 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号